
Cardiovascular Systems Biology
Through High-resolution Mass Spectrometry-based Comparative Proteomics and Metabolomics
Top-down Mass Spectrometry (MS)-based proteomics
|
Mass Spectrometry (MS)-based proteomics is a powerful tool for systems biology since it provides a systematic, global, unbiased, and quantitative assessment of proteins, including interactions, modifications, location, and function.1-4 Post-translational modifications (PTMs) modulate protein activity, stability, localization, and function, playing essential roles in many critical cell signaling events in both healthy and disease states.5 Dysregulation of a number of PTMs such as protein acetylation, glycosylation, hydroxylation, and phosphorylation, has been implicated in a spectrum of human diseases.6-9 The conventional peptide-based bottom-up shotgun proteomics approach is widely used but has intrinsic limitations for mapping protein modifications due to the dramatically increased complexity in examining an already complicated proteome as each protein is digested into many peptide components as well as loss of specific information concerning the protein since only a small and variable fraction of the digested peptides are recovered 10 (Figure 1). |
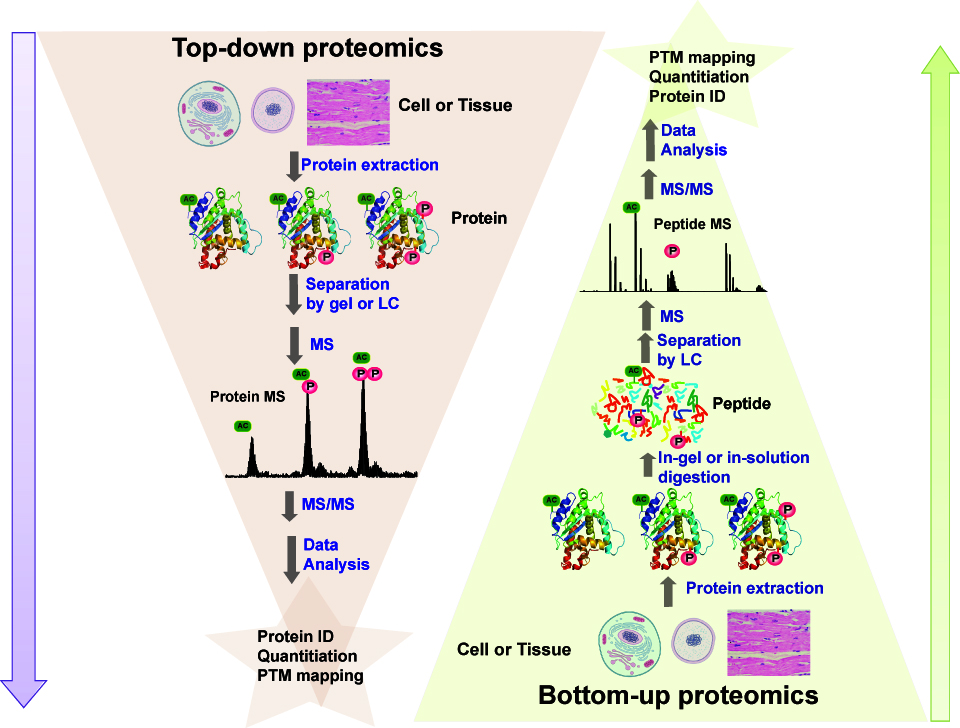 Figure 1. Schematic illustration of the difference between protein-based top-down and peptide-based bottom-up proteomics.6 |
In contrast, the protein-based top-down MS-based proteomics approach is arguably the most powerful technique for analysis of protein modifications.9,11 In the top-down approach, intact proteins are analyzed, which greatly simplifies sample preparation and reduces the mixture complexity as no proteolytic digestion is required (Fig. 1).12-17 Subsequently, specific proteins of interests can be “gas-phase” purified and modification sites can be mapped by tandem MS (MS/MS) strategies. The top-down MS provides comprehensive sequence information for the whole protein by detecting all types of PTMs (e.g. phosphorylation, proteolysis, acetylation) and sequence variants (e.g. mutations, polymorphisms, alternatively spliced isoforms) simultaneously in one spectrum (a "bird's eye view”) without a priori knowledge.11 We have made significant advances in top-down MS for analysis of large intact proteins purified from complex biological samples including cell and tissue lysate as well as body fluids.15,18-20 We have shown that top-down MS has unique advantages for unraveling the molecular complexity, quantifying modified protein forms, deep sequencing of intact proteins, mapping modification sites with full sequence coverage, discovering unexpected modifications, identifying and quantifying positional isomers and determining the order of multiple modifications.11,19,21-23 Moreover, we have shown that a tandem mass spectrometry technique, electron capture dissociation (ECD), is especially useful for mapping labile PTMs such as phosphorylation which are well-preserved during the ECD fragmentation process.18,21 Notably, we have been able to isotopically resolve large proteins (>115 kDa) with very high mass accuracy (1-3 ppm) and extended ECD to characterize very large phosphoproteins (>140 kDa) (Figure 1).15
Nevertheless, the top-down MS approach still faces significant challenges in terms of protein solubility, separation, and the detection of low abundance and large proteins, as well as under-developed data analysis tools.6,9 Consequently, new technological developments are urgently needed to advance the field of top-down proteomics. We have been establishing an integrated top-down disease proteomics platform to globally examine intact proteins extracted from tissues for the identification and quantification of proteins and possible PTMs present in vivo. Specifically, we are developing novel approaches to address the current challenges in top-down MS-based proteomics (Table 1 ). |
 Table 1. Novel approaches to address the challenges in top-down proteomics. Table 1. Novel approaches to address the challenges in top-down proteomics. |
A. To address the protein solubility challenge, we are developing new degradable surfactants that can effectively solubilize proteins and are compatible with top-down MS. we have recently developed an MS-compatible slowly degradable Surfactant (MasDeS) that can effectively solubilize proteins.24 Furthermore, we demonstrated that the solubility of membrane protein was significantly improved with the addition of this new surfactant. We are also developing different types of degradable surfactants and evaluating their performance for top-down proteomics.
B. To address the proteome complexity challenge, we are developing new chromatography materials and novel multi-dimensional liquid chromatography (MDLC) strategies to separate intact proteins. To address the proteome complexity challenge, we are developing new chromatography materials and novel strategies for multi-dimensional liquid chromatography (MDLC) to separate intact proteins. We have demonstrated the use of ultrahigh pressure size exclusion chromatography (UHP-SEC)25 and hydrophobic interaction chromatography (HIC)26 for top-down proteomics. Moreover, we have developed a novel 3DLC strategy by coupling HIC with ion exchange chromatography (IEC) and reverse phase chromatography (RPC) for intact protein separation.27 We demonstrated that this 3D (IEC-HIC-RPC) approach greatly outperformed the conventional 2D IEC-RPC approach. We are now developing novel chromatography materials for intact protein separation.
C. To address the proteome dynamic range, we have been developing novel nanomaterials that can bind low abundance proteins with PTMs (e.g. phosphorylation) with high specificity in collaboration with a nanotechnologist, Prof. Song Jin (U. of Wisconsin).28,29 The current focus is to develop multivalent nanoparticle (NP) reagents for capturing phosphoproteins globally out of the human proteome for top-down MS analysis of intact phosphoproteins.30
D. To address the challenge in under-developed software, we are developing user-friendly and versatile software interface for comprehensive analysis of high-resolution top-down MS-based proteomics data. Previously, we have developed MASH Suite, a versatile and user friendly software interface for processing, interpreting, visualizing and presenting high resolution MS data.31 Recently, we have developed MASH Suite Pro, a comprehensive, user-friendly and freely available program tailored for top-down high-resolution mass spectrometry (MS)-based proteomics (Manuscript submitted). MASH Suite Pro significantly simplifies and speeds up the processing and analysis of top-down proteomics data by combining tools for protein identification, quantitation, characterization, and validation into a customizable and user-friendly interface.
We envision that by taking this multi-pronged approach to overcome the challenges facing top down proteomics, we will significantly advance the burgeoning top-down proteomics field, which recently gained momentum through the creation of the Consortium for Top-down Proteomics (http://www.topdownproteomics.org/).
References:
- Cravatt, B. F.; Simon, G. M.; Yates, J. R. Nature 2007, 450, 991-1000.
- Cox, J.; Mann, M. Annual Review Biochemistry 2011, 80, 273-299.
- Aebersold R.; Mann, M. Nature 2003, 422, 198-207.
- Altelaar, A. F. M.; Munoz, J.; Heck, A. J. R. Nature Review Genetics 2013, 14, 35-48.
- Mann, M.; Jensen, O. N. Nature Biotechnology 2003, 21, 255-261
- Gregorich, Z. R.; Chang, Y. H.; Ge, Y. Pflugers Archiv-European Journal of Physiology 2014, 466, 1199-1209.
- Krueger, K. E.; Srivastava, S. Molecular & Cellular Proteomics 2006, 5, 1788-1810.
- Karve, T. M.; Cheema, A. K. J. Amino Acids 207691
- Gregorich, Z. R.; Ge, Y. Proteomics 2014, 14, 1195-1210.
- Chait, B. T. Science 2006, 314, 65-66
- Zhang, H.; Ge, Y. Circulation-Cardiovascular Genetics 2011, 4, 711
- Kelleher, N. L.; Lin, H. Y.; Valaskovic, G. A.; Aaserud, D. J.; Fridriksson, E. K.; McLafferty, F. W. Journal of the American Chemical Society 1999, 121, 806-812
- Ge, Y.; Lawhorn, B. G.; ElNaggar, M.; Strauss, E.; Park, J. H.; Begley, T. P.; McLafferty, F. W. Journal of the American Chemical Society 2002, 124, 672-678
- Kelleher, N. L. Analytical Chemistry 2004, 76, 196A-203A
- Ge, Y.; Rybakova, I. N.; Xu, Q.; Moss, R. L. Proceedings of the National Academy of Sciences of the United States of America 2009, 106, 12658-12663
- Siuti, N.; Kelleher, N. L. Nature Methods 2007, 4, 817-821
- Han, X. M.; Jin, M.; Breuker, K.; McLafferty, F. W. Science 2006, 314, 109-112
- Ayaz-Guner, S.; Zhang, J.; Li, L.; Walker, J. W.; Ge, Y. Biochemistry 2009, 48, 8161-8170
- Zhang, J.; Dong, X.; Hacker, T. A.; Ge, Y. Journal of the American Society for Mass Spectrometry 2010, 21, 940-948
- Peng, Y.; Chen, X.; Sato, T.; Rankin, S. A.; Tsuji, R. F.; Ge, Y. Analytical Chemistry 2012, 84, 3339-3346
- Zabrouskov, V.; Ge, Y.; Schwartz, J.; Walker, J. W. Molecular & Cellular Proteomics 2008, 7, 1838-1849
- Kuhn, P.; Xu, Q.; Cline, E.; Zhang, D.; Ge, Y.; Xu, W. Protein Science 2009, 18, 1272-1280
- Zhao, D. S.; Gregorich, Z. R.; Ge, Y. Analytical Chemistry 2013, 13, 3256-3260
- Chang, Y.; Gregorich Z. R.; Chen, A. J.; Hwang L.; Guner, H.; Yu, D.; Zhang, J.; Ge, Y. Journal of Proteome Research 2015, 14, 1587-1599
- Chen, X.; Ge, Y. Proteomics 2013, 13, 2563-2566
- Xiu, L.; Valeja, S. G.; Alpert, A. J.; Jin, S.; Ge, Y. Analytical Chemistry 2014, 86, 7899-7966
- Valeja, S. G.; Xiu, L.; Gregorich, Z. R.; Guner, H.; Jin, S.; Ge, Y. Analytical Chemistry 2015, 87, 5363-5371
- Nelson, C. A.; Szczech, J. R.; Dooley, C. J.; Xu, Q.; Lawrence, M. J.; Zhu, H.; Jin, S.; Ge, Y. Analytical Chemistry 2010, 82, 7193-7201
- Nelson, C. A.; Szczech, J. R.; Xu, Q.; Lawrence, M. J.; Jin, S.; Ge, Y. Chemical Communications 2009, 6607-6609
- Hwang, L.; Ayaz-Guner, S.; Cai, W.; Gregorich Z. R.; Jin, S.; Ge, Y. Journal of the American Chemical Society 2015, 137, 2432-2435
- Guner, H.; Close, P. L.; Cai, W. X.; Zhang, H.; Peng, Y.; Gregorich, Z. R.; Ge, Y. Journal of the American Society for Mass Spectrometry 2014, 25, 464-470
Sponsors
|
|
|
|
|